MHCquant: Identify and quantify peptides from mass spectrometry raw data
MHCquant is an nf-core best-practice bioinformatics analysis pipeline used for quantitative processing of data dependent (DDA) peptidomics data.
It was specifically designed to analyze immunopeptidomics data, which deals with the analysis of affinity-purified, unspecifically cleaved peptides presented on major histocompatibility complex (MHC) molecules.
This analysis has central implications for clinical research and T cell-centric immunotherapies in the context of cancer vaccines and personalized medicine.
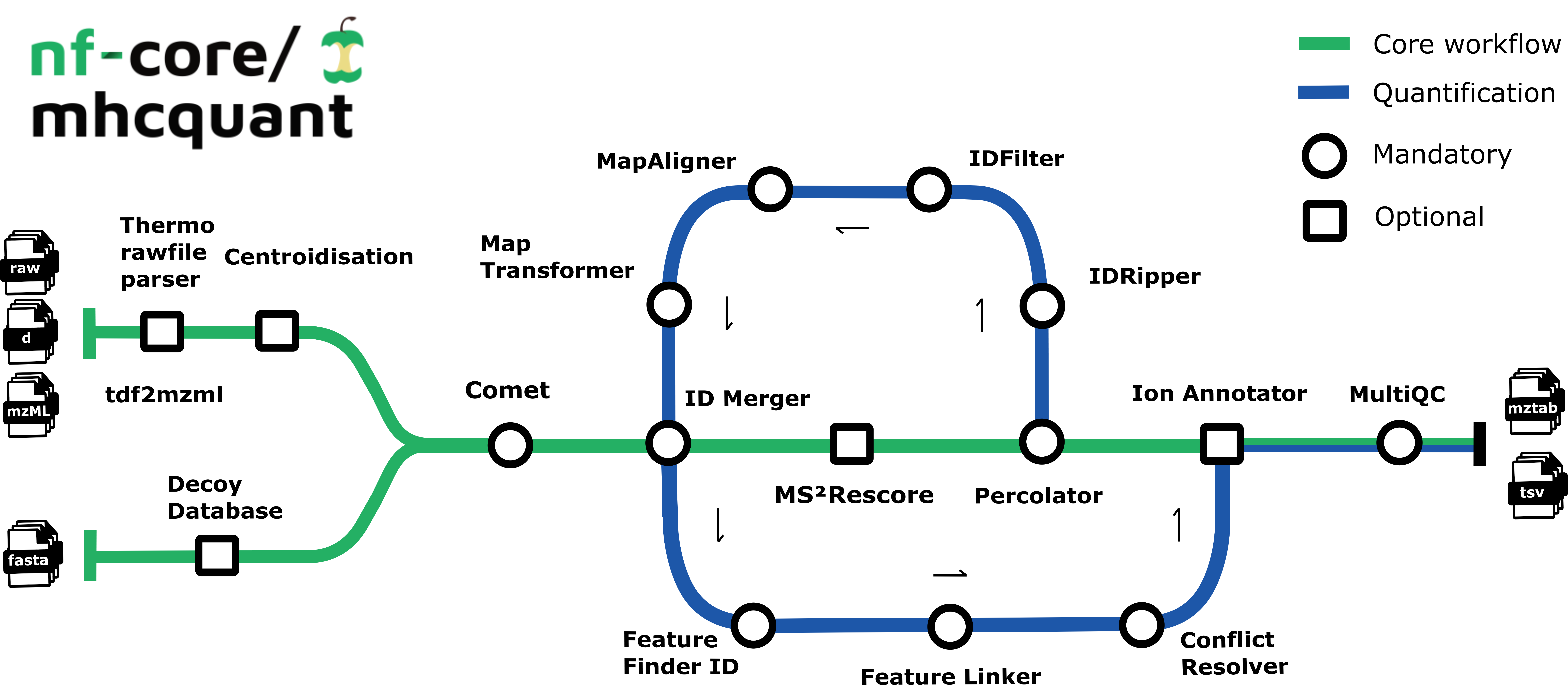
The workflow is based on the OpenMS C++ framework for computational mass spectrometry. Spectrum files (mzML/Thermo raw/Bruker tdf) serve as inputs and a database search (Comet) is performed based on a given input protein database. Peptide properties are predicted by MS²Rescore, which combines retention time prediction (DeepLC), fragment ion intensity prediction (MS²PIP), and ion mobility prediction (Ionmob) to dramatically boost immunopeptide identification rates. FDR rescoring is then applied using Percolator or Mokapot based on a competitive target-decoy approach (reversed decoys). The pipeline supports both local FDR control (per sample-condition group) and global FDR control (across all samples), providing flexibility for different experimental designs.
Moreover, binding predictions on specified alleles using various binding prediction tools can be directly run on all matched PSMs to directly annotate MHC binders and non-binders and calculate a refined FDR on a subset of PSMs leading to higher identification rates. For label-free quantification, all input files undergo identification-based retention time alignment and targeted feature extraction. The pipeline can also generate spectrum libraries suitable for DIA-based searches as well as computing consensus epitopes using epicore.
In addition, mutation-informed fastas can be specified to search for patient-specific neo-epitopes.
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It comes with docker / singularity containers making installation trivial and results highly reproducible.
The Nextflow Workflow is available at:
https://github.com/nf-core/mhcquant
Citation
If you use MHCquant, please cite:
Scheid et al. (2025). MHCquant2 refines immunopeptidomics tumor antigen discovery. Genome Biology.